Scientists Present Self-Learning Interatomic Potential for Paramagnetic Materials
June 23, 2025 -- An international team of researchers has developed a method to tune a mathematical model of magnetic interactions that helps to realistically simulate and design materials with desired properties and predict their behavior before experimental testing. The research was published in Physical Review B.
Recently, much research has been focused on machine-learning interatomic potentials, which can ensure fast and accurate simulation of a material’s structure and properties. Quantum-mechanical methods, such as density functional theory, are very accurate but too time-consuming and computationally expensive. Machine learning accelerates the computations for large systems while offering virtually the same level of accuracy. However, physical reliability remains a major issue in machine learning applications.
In their new study, researchers from Skoltech, MIPT, and HSE University in collaboration with their international colleagues proposed an automatic learning algorithm for a machine-learning interatomic potential with magnetic degrees of freedom to accelerate the lengthy quantum-mechanical calculations in studying of paramagnetic materials without compromising accuracy.
Magnetic moments represent a new variable, which complicates the potential’s training. The simulation with a magnetic interatomic potential consists of two steps. The first step aims to optimize magnetic moments with fixed atomic coordinates and lattice parameters in order to minimize the system’s total energy. The second step involves molecular dynamics simulation, where magnetic moments are fixed while atomic coordinates and lattice parameters are varied based on magnetic interactions.
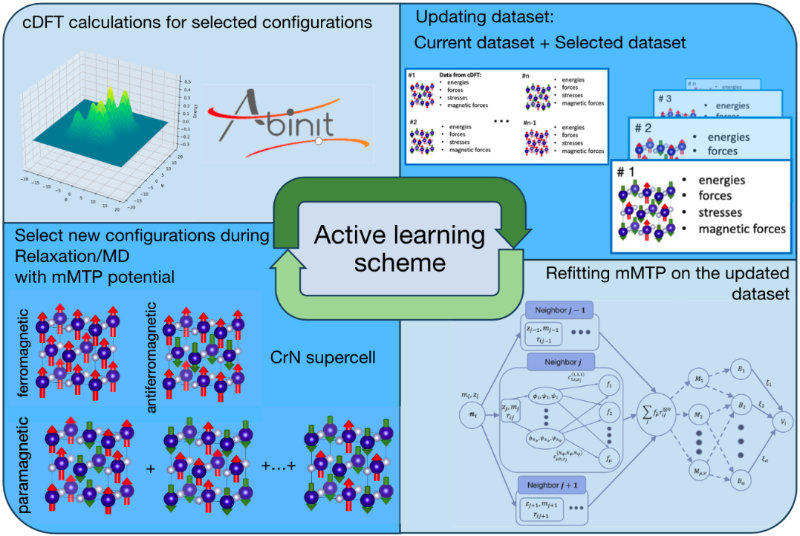
The training is also complicated by the magnetic moments present in the potential’s functional form. To solve this issue, the team developed an algorithm that automatically selects optimal configurations for the training dataset. The algorithm identified the configurations generated during the simulation and ran density functional theory calculations for the selected configurations. The results were then added to the training dataset.
“The standout feature of our potential is the ability to select configurations during the simulation, including the molecular dynamics simulation step. Since the potential is able to pick relevant configurations for further DFT simulation and refitting, the training dataset can be compiled automatically. The potential also takes into account the magnetic moments of the configurations selected during active learning,” said Ivan Novikov, an associate professor at the HSE Faculty of Computer Science, an associate professor at the MIPT Department of Chemical Physics of Functional Materials, and a senior research scientist at Skoltech AI.
Here is a detailed breakdown of how active learning of an interatomic potential with magnetic degrees of freedom proceeds (see image). The training starts with an initial potential and a training dataset. It is followed by active learning during the search for equilibrium magnetic moments, relaxation or MD simulation. If the degree of extrapolation is too high for any particular configuration, the simulation is stopped and new configurations are selected from those already generated. Then, density functional theory simulation is performed with magnetic moment constraints for the selected configurations. The process ends with dataset updating and potential refitting. It is then repeated from the beginning until no configuration is preselected during the equilibration/relaxation/molecular dynamics simulation step.
The researchers tested their new approach on chromium nitride, a material with a cubic crystal lattice similar to that of cooking salt. Chromium nitride, whose properties are well known, helped confirm the reliability of the method. However, this material is also known to be paramagnetic above room temperature, which was an additional hindrance to the validation. The tests revealed that the algorithm precisely reproduced the elastic constants and thermal properties. The calculated phonon spectra were also consistent with the experimental data. According to the researchers, the algorithm is universal and could be applied for other materials.
Thus, the proposed approach proved to be highly accurate in reproducing the mechanical, dynamic, and thermal properties of paramagnetic chromium nitride, while demonstrating opportunities for its wide application in materials engineering.
“We plan to add noncollinear magnetism to our potential’s functional form. We also want to develop and test a method for predicting the paramagnetic transition temperature using the Monte Carlo technique with magnetic moments flipping during molecular dynamics simulations,” Novikov said.
The study involved researchers from Skoltech, MIPT, HSE University, the Institute of Solid State Chemistry and Mechanochemistry of SB RAS, Emanuel Institute of Biochemical Physics of RAS, the University of Cagliari in Italy, and the Materials Center Leoben in Austria.
Most Recent










NewsFlash
Industry Pioneers
Editor's Picks
-

HRL Reports Self-Controlled Silicon Quantum Processor in Nature
-

Cleveland Clinic and IBM Researchers Create Quantum Machine Learning Framework to Predict Neoantigen Immune Response
-

Using a Molecule as a Quantum Sensor
-

Four Leading Quantum Companies Sign-On to ABQ-Net, the First Open-Access Quantum Network in the United States